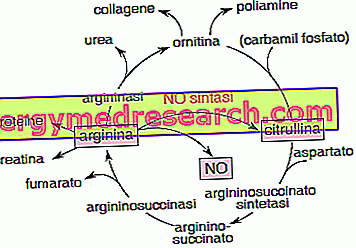
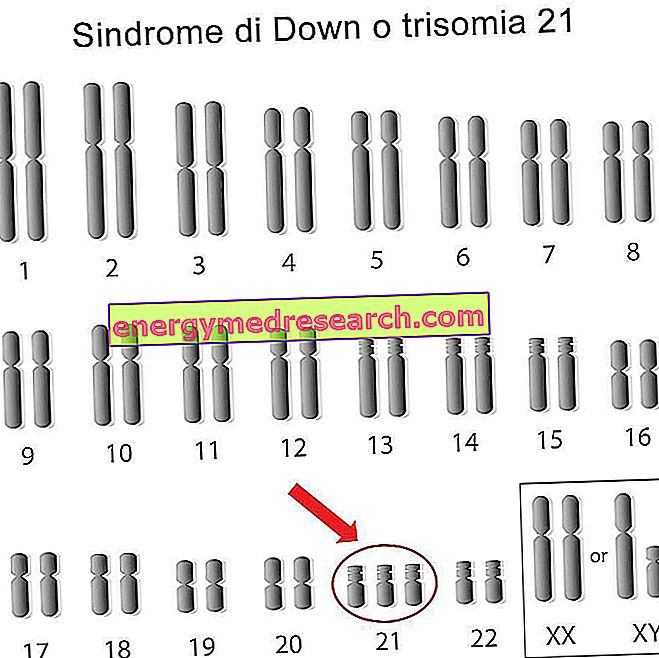
一般性 デュシェンヌ型筋ジストロフィー ( DMD )は、筋肉に影響を与える、遺伝性の疾患であり、遺伝することもあります。 患者は、基本的なタンパク質である ジストロフィン が存在しないために筋緊張を失います。 数年後には、筋肉の関与が非常に多くなるため、病気の患者は車椅子に入って呼吸を補助されます。 残念ながら、デュシェンヌ型筋ジストロフィーの治療法はありません。 現在の治療法は、症状を軽減し進行を遅らせることしかできません。 しかし、この試験は特定の治療法を見つけるために積極的です。 染色体 デュシェンヌ型筋ジストロフィーを理解するためには、ヒト染色体セットを説明する前提が必要です。 健康な人間のあらゆる細胞は23組の染色体を含んでいます。 これらの 染色体 のペア は性的です 、すなわち、それは個人の性別を決定します。 残りの22対は、代わりに 常染色体で 構成されています。 それゆえ、全部で、ヒトゲノムは46の染色体を有する。 染色体の変化 染色体の各ペアは特定の 遺伝子を 含みます。 染色体に 突然変異 が発生すると、遺伝子に欠陥が生じる可能性があります。 この欠陥遺伝子は結果的に欠陥タンパク質を発現する。 それとは反対に、染色体の数が変化するとき、我々は 異数性 について話す。 この場合、染色体は2つではなく3つ(トリソミー)でも1つのみ(モノソミー)でもかまいません。